Mirdametinib (PD0325901): Modulating Endochondral Ossification
Mirdametinib (PD0325901) and Selumetinib: Modulating Endochondral Ossification via MEK Inhibition
Abstract
MEK inhibitors MEKi Mirdametinib (PD0325901) and AZD6244 Selumetinib are drugs currently under clinical investigation for cancer treatment, however the Ras–MAPK pathway is also an important mediator of normal bone cell differentiation and function. In this study we examined the effects of these compounds on endochondral processes using both in vitro and in vivo models. Treatment with PD0325901 or AZD6244 significantly increased Runx2 and Alkaline phosphate gene expression in calvarial osteoblasts and decreased TRAP positive cells in induced osteoclast cultures. To test the effects of these drugs on bone healing, C57/Bl6 mice underwent a closed tibial fracture and were treated with PD0325901 or AZD6244 at 10 mg/kg/day. Animals were culled at day 10 and at day 21 post-fracture for analysis of the fracture callus and the femoral growth plate in the contralateral leg. MEKi treatment markedly increased cartilage volume in the soft callus at day 10 post-fracture with 60 percent increase for PD0325901 and 20 percent increase for AZD6244 and continued treatment led to a delay in cartilage remodeling. At the growth plate, we observed an increase in the height of the hypertrophic zone relative to the proliferative zone of 78 percent in PD0325901 treated mice. Osteoclast surface was significantly decreased both at the terminal end of the growth plate and within the fracture calluses of MEKi treated animals. The mechanistic effects of MEKi on genes encoding cartilage matrix proteins and catabolic enzymes were examined in articular chondrocyte cultures. PD0325901 or AZD6244 led to increased matrix protein expression Col2a1 and Acan and decreased expression of catabolic factors Mmp13 and Adamts-5. Taken together, these data support the hypothesis that MEKi treatment can impact chondrocyte hypertrophy, matrix resorption, and fracture healing. These compounds can also affect bone architecture by expanding the hypertrophic zone of the growth plate and reducing osteoclast surface systemically.
Keywords Ras–MAPK, Fracture healing, Cancer, Cartilage remodeling, Endochondral ossification
Introduction
The Ras signaling molecule is an important mediator of cell survival, proliferation and function. The Ras–Raf–MEK–ERK axis also called the Ras–MAPK pathway has a central role in cell signaling, and is activated in many human cancers. Activating mutations in the Ras superfamily have been identified in approximately 15 percent of all human cancers, while B-Raf mutations affect a number of tissue specific cancers. MEK inhibitors plays a key role in transducing these upstream signals, and has thus been a highly studied therapeutic target.
MEK inhibitors MEKi PD0325901 Pfizer and AZD6244 Selumetinib, ARRY-142886, AstraZeneca are small molecule drugs that can reduce tumor growth in preclinical xenograft models. These compounds are undergoing clinical trials for treatment of a variety of tumor types with PD0325901 being used for advanced stage cancers Trial ID NCT01347866, and AZD6244 being used in over 50 trials in oncology patients. To date, the adverse effect profile has been limited to nausea, edema, skin rashes, visual disturbances, fatigue and diarrhea, which have been manageable and dose dependent. The development of specific and bioavailable MEKi compounds AZD6244 Selumetinib and PD0325901 provides a unique opportunity to explore the role of MEK signaling in skeletal biology. The potential impact of MEKi in bone is of particular relevance to cancer patients as these treatments can lead to bone loss and increased fracture risk. In breast cancer patients without bone metastasis or MEKi treatment, the incidence of vertebral fractures was 5 fold greater than the normal population. In women with recurrent breast cancer, the risk was 20 fold greater than normal. As these drugs progress through clinical development, a better understanding of their skeletal manifestation is important. Three registered clinical trials are focused on pediatric patients, yet limited information is available regarding the role of MEKi on the developing skeleton, skeletal homeostasis, and bone repair.
Endochondral ossification is a key process involved with both skeletal development and repair. During development, mesenchymal progenitors are recruited and condense to differentiate into pre-chondrocytes. Differentiation of pre-chondrocytes to chondrocytes is regulated by the master regulator gene Sox9, in addition to Sox4 and Sox5. During this early phase of differentiation, chondrocytes express collagen type I Colla1, and then secrete increasing amounts of collage type II Col2a1 and aggrecan Acan. As chondrocytes continue through the cell cycle, they begin to organize into columns of proliferating chondrocytes, and eventually enter the terminal phase of differentiation and become hypertrophic chondrocytes. Hypertrophic chondrocytes secrete high levels of collagen type X Col10a1, vascular endothelial growth factor Vegf, and in the final stages of differentiation, matrix metalloproteinase 13 Mmp13. The expression of Mmp13 and Vegf promote cartilage remodeling and invasion of vessels that enable infiltration of osteoblasts and osteoclasts. In the final stages of endochondral ossification, the invasion of bone cells replaces the cartilage with stronger trabecular bone.
Endochondral ossification is a fundamental process that occurs during development at the growth plates of all long bones, and contributes to bone lengthening. This process is also recapitulated during fracture healing. Fracture healing follows a sequential series of events: the initial inflammatory reaction, the development of a cartilaginous soft callus, followed by laying down of trabecular bone of the hard callus by endochondral ossification, finally, the trabecular bone is remodeled into stronger lamellar bone that takes the shape of the original bone. In mice, the cartilage of the soft callus peaks at about 10 days, and the trabecular bone of the hard callus peaks at about 21 days post fracture.
Genetic mouse models have demonstrated the importance of the Ras–MAPK pathway in chondrocyte and osteoblast cell differentiation and function. Transgenic mice with inactivating or overactive mutations of MEK driven by the osteocalcin promoter have yielded important insights. These studies intimated an important role for MEK in promoting the bone forming activity of late stage osteoblasts where osteocalcin is expressed. In contrast, inactivation of ERK1/2 the downstream targets of MEK using the Prx1 promoter expressed in mesenchymal progenitors of the limb promoted chondrogenesis at the expense of osteogenesis. Intriguingly, this mouse model showed decreased RANKL expression in the humerus and tibia, indicating decreased resorptive signaling. Furthermore, animal studies have identified that ERK is highly expressed in hypertrophic chondrocytes, suggesting an important regulatory role. Taken together, these studies suggest that the Ras–MAPK pathway has an important role in the specification and differentiation of osteochondral progenitors as well as the function of mature osteoblasts.
In this study we have evaluated the impact of AZD6244 and PD0325901 on cultured cells, fracture healing, and in the growth plates of mice. MEKi treatments were applied in vitro to osteoblasts, chondrocytes, and osteoclasts. To address the role of MEK inhibition on bone repair, mice underwent closed fracture surgery and were treated with PD0325901 and AZD6244 at 10 mg/kg/day using several dosing regimens. Fracture specimens were analyzed by microCT and tissue histology. To determine how these compounds may impact bone growth, the growth plates of contralateral limbs were also analyzed. Biochemical markers for bone turnover were also assessed.
Materials and Methods
Calvarial Osteoblast Cultures Calvarial cultures were generated from neonatal mice harvested within one week of birth as previously published. Briefly, mice were decapitated and the calvaria were washed in Dulbecco’s PBS while stripping any remaining fibrous tissue. Calvaria were minced and digested three times in Collagenase D at 1 mg/ml Roche in αMEM Invitrogen at 37 degrees Celsius for 15 min per digestion. The resulting calvaria fragments were plated in 10 percent fetal bovine serum in αMEM with penicillin/streptomycin and cells were allowed 3 to 5 days to grow onto the plate before splitting for experiments. At confluence, cultures were differentiated with α-MEM containing 10 percent FBS and penicillin/ streptomycin, supplemented with 10 mM β-glycerophosphate, 50 microgram per ml ascorbic acid, and 100 ng/ml of rhBMP-2 Medtronic Australasia. PD0325901 was dissolved in DMSO and added to the differentiation media.
Induction of mineralization was established by staining for calcium deposits in the well. Cells were fixed with 4 percent paraformaldehyde for 10 min, followed by 5 minute incubation with Alizarin Red S 40 mM, pH 4.2. Samples were washed three times with water and mineralization nodules were quantified using a light microscope.
To examine osteogenesis, quantitative PCR was performed on a Rotorgene LightCycler. RNA was harvested from cells using Trizol Reagent Invitrogen, and cDNA using Superscript III Invitrogen. qPCR was performed using a SYBR Green Mastermix Stratagene using the following primer sequences: ALP-F 5′-GGGACTGGTACTCGGATAACGA-3′, ALP-R 5′-CTGATATGCGATGTCCTTGCA-3′; Runx2-F 5′-AGCCTCTTCAGCGCAGTGAC–Runx2-R 5′-CTGGTGCTGCGATCCCAA. Dissociation curves using these primer pairs resulted in a single amplification peak, confirming the specificity of these primers.
Osteoclast Cultures Bone marrow was harvested from the femur of C57/Bl6 mice aged to 8 to 10 weeks. The bone marrow suspension was filtered using a 40 micrometer filter, washed in PBS, and resuspended in DMEM with 10 percent FBS and antibiotics. M-CSF 20 ng/ml; R&D Systems, MN, USA and RANKL 100 ng/ml; R&D Systems, MN, USA were used to differentiate the cells towards the osteoclastic lineage, and PD0325901 or AZD6244 were co-delivered as appropriate in the media. Cells were incubated and differentiated for 8 days before performing a TRAP stain to assess osteoclast numbers in the well as previously described.
Surgical Methods Female C57Bl6 mice aged approximately 11 weeks were used in closed fracture surgery experiments. All fractures were generated at the tibial mid-shaft near the fibular junction by experienced staff based on our published methodology. Anesthesia was induced with ketamine 35 mg/kg and xylazine 4.5 mg/kg via intraperitoneal injection with a 27-G needle and maintained using inhaled isoflurane. Next a small incision was made slightly below the knee and an entry point made using a 27-G needle. An intramedullary rod a 30-G needle or a 0.3 mm-diameter stainless steel insect pin was surgically inserted inside the medullary canal of the tibia, and a second pin inserted for stability. A non-comminuted, transverse fracture near the fibular junction was confirmed via radiography using a digital X-ray machine Faxitron X-ray Corp., Wheeling, IL. The wound was closed using 5–0 nylon suture Ethicon Inc., Somerville, NJ. The mice were allowed to recover on a heated pad and then placed in recovery cages. Pain was managed using buprenorphine 0.05 mg/kg subcutaneously postoperatively, then every 12 h as required and dehydration was controlled via post-operative i.p. injection with 1 ml sterile saline. Fracture repair was monitored by weekly radiography Faxitron X-ray and in the event where internal fixation had failed due to pin slippage, bending or breakage the affected mouse was culled and excluded from subsequent analysis. At the experimental endpoints, animals were euthanized using carbon dioxide and specimens collected post-mortem for radiography and histology. All animal experiments were undertaken with approval from the local animal ethics committee.
Treatment with PD0325901 or AZD6244 Mice were single housed prior to the beginning of the experiment, and they were trained to eat a strawberry jelly as drug vehicle. This jelly was composed of 0.8 percent DMSO Sigma-Aldrich, St. Louis, MO, USA, 16 percent Splenda® Splenda® Low Calorie Sweetener, Johnson-Johnson Pacific Pty, NSW, Australia, 9.6 percent gelatine Davis Gelatin, GELITA Australia Pty, NSW, Australia and 7.9 percent flavoring QUEEN Flavoring Essence Imitation Strawberry, Queen Fine Foods Pty, QLD, Australia. The jelly was given to the mice with or without PD0325901 Selleck Chemicals, TX, USA or AZD6244 Chemietek, IN, USA dissolved in DMSO. This method of administration proved very effective at drug delivery, and reduced the stress normally associated with oral gavage. Single-housing of the mice assured that the jelly was being consumed at the appropriate dose. This method has been previously described.
Histological Analysis Fractured tibiae were removed along with the surrounding soft tissues, fixed overnight in 4 percent paraformaldehyde, and then stored in 70 percent alcohol at 4 degrees Celsius prior to decalcification. For growth plate analyses, the contralateral femora were removed and processed in a similar fashion. Bone samples were decalcified in 0.34 M EDTA pH 8.0 for 21 days at 4 degrees Celsius on shaker with changes every 2 to 3 days. Samples were embedded in paraffin blocks and 5 micrometer-thick sections were cut and stained with Picrosirius Red/Alcian Blue to stain for bone and cartilage, and counterstained with Harris Hematoxylin. Adjacent sections were stained for tartrate-resistant acid phosphatase TRAP to highlight osteoclasts, and counterstained with 0.4 percent Light Green. Stained sections were quantified and analyzed using Bioquant Analysis System Nashville, TN, USA. One section per biological sample was analyzed, and for histology, the values were normalized to tissue area. These methods have been previously published.
Serum RANKL and OPG Blood was collected via cardiac puncture of C57/B16 animals at the experimental endpoint. Blood was allowed to clot for 30 min at room temperature before centrifugation. The serum was stored at minus 20 degrees Celsius. For RANKL and OPG readings, blood samples were freshly thawed and assayed using the RANKL or OPG kits supplied by R&D Systems Minneapolis, USA.
Sheep Articular Chondrocyte Cultures Under sterile conditions full depth articular cartilage was removed from the femoral and tibial surfaces of skeletally mature 2 to 3 year old ovine knee joints within 6 h of sacrifice. Chondrocytes were isolated by digesting cartilage in 0.1 percent Pronase Roche in Dulbecco’s modified Eagle’s medium – Nutrient mixture F-12 Ham DMEM/F12; Sigma with 10 percent volume per volume fetal calf serum FCS; Ausgenex, Brisbane, Australia for 2 h at 37 degrees Celsius. Tissue was then washed and digested with 0.05 percent collagenase Sigma in DMEM/F12 with 10 percent FCS overnight at 37 degrees Celsius with agitation. Cells were then cultured replicate cultures for each analytical point in fresh media with or without interleukin-1-alpha IL-1; 1 ng/mL; PeproTech in the presence or absence of PD0325901 or AZD6244 1 nM, 100 nM for 24 h before RNA harvest using Trizol Reagent. This method is previously described.
Statistical Methods Sample size for each experiment is indicated in the appropriate figure legend. Samples that were larger than two standard deviations were defined as outliers and not included in the analysis. For fracture studies, a Fisher Exact Test was performed based on unions and non-unions for the treatment and untreated groups. For the histological analysis, a one-way analysis of variance was performed, with a Dunnett’s or Tukey’s multiple comparisons post-test. For quantitative PCR, a t-test was used to determine significant between individual genes. All analyses were performed using GraphPad Prism 6 La Jolla, CA, USA.
Results
MEK Inhibition Promotes Osteoblast Differentiation and Suppresses Osteoclast Formation in Vitro To determine the effect of MEK inhibition on osteoblasts, primary calvarial osteoblasts were cultured in the presence of the potent bone anabolic factor rhBMP2 100 ng/ml and two different MEKi compounds, PD0325901 and AZD6244 Selumetinib. Cells were treated for nine days, and RNA was harvested to examine the expression of Runx2 and Alkaline phosphatase Alp, two important osteoblastic markers. Quantitative PCR for Runx2 and Alp expression revealed that PD0325901 and AZD6244 were able to promote osteoblastogenesis in a dose dependent manner at 10 nM and 100 nM. Additionally, cultures were stained with Alizarin Red S for mineralized nodules, which were subsequently quantified. Four days after treatment with PD0325901, we saw a large and significant increase in the number of mineralized nodules compared to DMSO vehicle. At this time point and concentration, AZD6244 did not impact mineralization suggesting it may be less potent than PD0325901.
To understand the role of MEK in osteoclastogenesis, hematopoietic progenitors from the bone marrow were differentiated in the presence of recombinant MCSF and RANKL. Cells were incubated with AZD6244 or PD0325901 at 1 nM, 10 nM, and 100 nM, and cultured for 8 days. Osteoclast differentiation was quantified by TRAP stain. MEK inhibition using either compounds led to a decrease in the number of TRAP positive osteoclasts. PD0325901 showed a greater potency than AZD6244, with 100 nM AZD6244 reducing osteoclasts with a comparable efficacy to 10 nM of PD0325901.
Taken together, these data suggest that MEKI treatment can play an important role in bone formation and bone resorption. Therefore, in the following series of experiments, we evaluated the role of these inhibitors in fracture healing.
MEK Signals Modulate Progenitor Differentiation in Endochondral Fracture Repair C57B16 mice underwent a closed fracture surgery on the tibia, and PD0325901 or AZD6244 was administered orally at 10 mg/kg/day during the different stages of fracture healing. No adverse effects were reported at this dose, and mouse weight was not affected. Mice were treated in the 1 early phase of repair during hematoma and soft callus formation; in the 2 late phase during establishment of the hard callus and its subsequent remodeling; or 3 continuously throughout the healing process. Animals were harvested 10 and 21 days post fracture for analysis of the femur, fracture callus, and serum biochemistry. Animals harvested at day 10 post fracture received a total of 13 doses, while animals in the continuous group received 24 doses by day 21 time point.
Fractures were analyzed by microCT 21 days post-fracture to quantify the mineralized bone volume in the fracture callus. Continuous treatment with either PD0325901 or AZD6244 resulted in a significant increase in BV/TV bone volume relative to tissue volume. With PD0325901 the total callus BV bone volume was increased, but not significantly so; in AZD6244 treated animals the callus BV showed a 20 to 23 percent reduction p less than 0.01, but this was also associated with reductions in TV tissue volume of 27 to 32 percent p less than 0.05. To further analyze the calluses, detailed histology was assessed at day 10 and day 21 post-fracture.
Day 10 specimens revealed a significantly greater cartilage area when treated with PD0325901 compared to vehicle controls. Treatment with AZD6244 also produced a trend towards increased cartilage but this did not reach statistical significance. This time point represents the peak cartilage formation step during fracture healing, suggesting early MEKi treatment promoted the accumulation or formation of cartilage. Day 21 specimens similarly showed considerable and statistically significant retention of cartilage with continuous MEKi treatment with either PD0325901 or AZD6244.
Mice treated with MEKi after the establishment of the cartilaginous soft callus Late group, days 10 to 21 showed an increase in the amount of cartilage remaining. This cartilage appeared characteristically hypertrophic and mineralized. The effects on cartilage retention of d10 to 21 treatment were more pronounced with PD0325901 treatment than with AZD6244. In contrast, cessation of MEKi treatment after day 10 Early group, days minus 3 to 10 led to negligible residual cartilage remaining by day 21. These data indicate that MEK inhibition may impact chondrocyte terminal differentiation and cartilage resorption.
MEK Inhibition Reduces Osteoclastogenesis in the Fracture Callus In fracture healing, osteoclasts are critical for remodeling of the hard callus, effects of MEKi on osteoclastogenesis were hypothesized based on cell culture data in vitro. A stain for tartrate-resistant acid phosphatase TRAP for osteoclasts was performed and significant effects were seen at 21 days with MEKi treatment. Treatment with PD0325901 Continuous resulted in a 43 percent decrease in osteoclast surface p equals 0.03. These effects were reversible, as PD0325901 Early showed no difference. Mice treated with AZD6244 Continuous also showed a significant 21 percent decrease in osteoclast surface p equals 0.005. However, unlike PD0325901 Early, AZD6244 Early showed a persistent decrease even after 11 days. This was not associated with significant changes in BV or BV/TV in the bone below the growth plate as measured by microCT data not shown.
Serum was collected from animals treated with AZD6244 at day 10 and at day 21 post-fracture and analyzed for RANKL and OPG biochemistry. 10 days of treatment with 10 mg/kg of AZD6244 produced a 42 percent reduction in circulating RANKL and no change in OPG. Intra-mouse variability was high and this difference did not reach statistical significance. 21 days of AZD6244 treatment led to a statistically significant reduction in RANKL 25 percent, P less than 0.05, but also comparable reduction in OPG. These data suggest the changes in osteoclast surface at the fracture site may reflect differences in the local fracture microenvironment rather than systemic changes in bone resorption.
MEK Inhibition Maintains Hypertrophic Chondrocytes at the Growth Plate The growth plates from the animals that underwent fracture surgery were also analyzed to help elucidate what stage of cellular differentiation MEKi treatment may influence. The specimens examined were either treated with MEKi for 13 days, 24 days, or 13 days with a wash-out period of 11 days. Growth plates were analyzed by comparison of the proliferative zones and hypertrophic zones. The hypertrophic zone was significantly expanded with 13 or 24 days PD0325901 treatment. The proliferative zone was correspondingly decreased, leaving the total growth plate length unchanged with treatment. After the 11 day washout period, no difference in hypertrophic zone was seen, suggesting the effects were reversible over the time period studied. In contrast, AZD6244 was not found to significantly affect the growth plate.
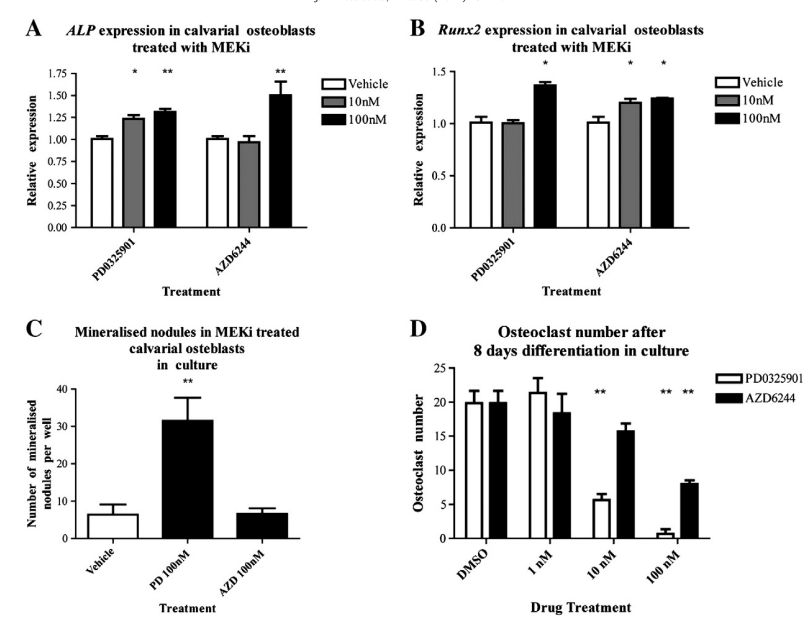
Fig. 1. MEK inhibition in calvarial osteoblasts and bone marrow derived osteoclasts. Calvarial osteoblasts were differentiated with rhBMP2 (100 ng/ml) and PD0325901 or AZD6244. RNA was harvested at day 9 and expression of Alkaline phosphatase and Runx2 was quantified (A, B). Cells were then stained with Alizarin Red S for mineral and quantified (C). MEKi treatment led to an increase in osteogenic potential. Bone marrow osteoclasts were differentiated using M-CSF and RANKL and co-treated with PD0325901 or AZD6244 for 8 days (D). A TRAP stain for osteoclasts revealed that MEKi treatment reduced osteoclast numbers in the well. (*p b 0.05, **p b 0.01, ANOVA).
To determine whether osteoclasts were affected below the growth plate of these animals, a TRAP stain was performed in the primary spongiosa. In mice treated with PD0325901, osteoclast number and surface were significantly reduced after 13 and 24 days, but again was reversible with 11 day washout. In contrast, the trend towards reduced osteoclast number and surface did not reach significance in animals treated with AZD6244 except in 13 day treatment/11 day washout group. This suggests some underlying differences in impact on osteoclasts and reversibility between the two agents.
MEK Inhibition Affects Expression of Matrix Factors and Catabolic Enzymes in Cultured Chondrocytes Based on the fracture healing and growth plate data, it was hypothesized that the skeletal effects of MEKi may mechanistically involve effects on cartilage matrix production and breakdown. This was further examined in vitro using sheep articular chondrocytes treated with PD0325901 and AZD6244. These cells were selected because they can be induced to strongly increase expression of catabolic enzymes with interleukin treatment.
MEKi treatment with 100 mM PD0325901 and 100 mM AZD6244 led to significant increases in the cartilage-specific matrix genes Collagen-2 Col2a1 and Aggrecan Acan. Collagen-1 Col1a1, a marker of chondrocyte de-differentiation, was not affected by PD0325901 and more modestly increased by 100 mM AZD6244. Treatment of chondrocytes with IL-1 resulted in significant decreases in the gene expression of Col1a1, Col2a1, and Acan. MEKi treatment led to partial rescue of genes encoding matrix proteins, with the most potent effects on Col1a1.
Expression of cartilage degrading enzymes Adamts-4 a disintegrin and metalloproteinase with thrombospondin motifs-4, Adamts-5, and Mmp13 was quantified. MEK inhibition led to significant decreases in Adamts-5 and Mmp13 expression, even with 1 mM PD0325901 and 1 mM AZD6244. Consistent with the articular chondrocyte culture model, 1 ng/ml IL-1 treatment dramatically increased expression of genes encoding these enzymes, in some cases by several orders of magnitude. While Adamts-4/5 expression were unchanged or slightly increased by the MEKi compounds, Mmp13 expression was significantly decreased.
Overall, these data indicate that MEKi compounds can increase the expression of specific cartilage matrix genes while decreasing Mmp13 expression, which would favor the accumulation and retention of cartilage.
Discussion
The Effects of MEK Inhibition on Bone Cells Numerous in vitro studies have attempted to evaluate the role of the Ras–MAPK pathway in bone cells, although a clear consensus has not yet been reached. The use of older MEKi compounds such as PD98059 or U0126 have been a useful guide, but these compounds are less specific that PD0325901 or AZD6244. In C2C12 muscle cells and MC3T3 pre-osteoblastic cells, PD98059 were found to promote osteoblastogenesis synergistically with rhBMP2. Chondrogenic micromass cultures treated with PD98059 or U0126 show increased expression of Acan and Col2a1, suggesting that they promote chondrogenesis. Our in vitro experiments with these highly specific inhibitors indicate that the Ras–MAPK pathway is a negative regulator of osteoblast maturation. MEK inhibition led to a greater relative increase in chondrocyte gene expression markers than in osteoblasts markers. This suggests that chondrocytes are more sensitive to variations in Ras–MAPK activity than osteoblasts.
The Effects of MEK Inhibition on Bone Repair Fracture healing can be temporally divided into an inflammatory phase, a soft callus phase, a hard callus phase, and a remodeling phase, although these stages show some overlap. The bone repair process involves the coordination of multiple cell types to restore normal bone shape and function. Our data reveals that treatment with PD0325901 or AZD6244 can significantly impair the endochondral ossification in a fracture callus. Peak cartilage was increased with PD0325901, cartilage remodeling was decreased, and the number of osteoclasts in the callus was decreased. Treatment with AZD6244 did not result in a significant increase in peak cartilage at day 10, but at day 21, we observed a significant increase in cartilage, suggesting impairment of cartilage resorption. The effects of PD0325901 and AZD6244 treatments were examined by microCT, and differences in bone parameters with MEKi treatment were noted. Based on the histology, it is possible that retention of mineralized cartilage may underlie some of the changes in BV/TV detected using this modality.
Serum samples revealed that the RANKL to OPG ratio was not changed with MEKi treatment, despite a change in osteoclast surface/bone surface. This is likely because RANKL is secreted by osteocytes embedded in the matrix to regulate osteoclasts locally, rather than through systemic osteoclast regulation. While osteoclast surface/bone surface was significantly reduced, we did not observe a significant effect on osteoclast number/bone surface, and this may be due to a lack of statistical power for this parameter.
The process of cartilage remodeling during endochondral ossification is normally undertaken by osteoclasts as well as other cell types that secrete matrix metalloproteinases MMPs. Insight into the cells that can participate in endochondral remodeling have been elucidated by several recent studies. Treatment with the MMP inhibitor MM1270 led to the accumulation of cartilage in a rat fracture model. In contrast, treatment with bisphosphonates which abrogate osteoclast-mediated resorption or OPG which blocks osteoclast formation activity lead to increased bone mineral content due to delayed hard callus remodeling, but no delay in cartilaginous/soft callus remodeling. Analogously, Mmp9 or Mmp13 knockout mice that underwent a fracture surgery show delayed cartilage remodeling, and a persistent cartilaginous fracture callus. These experiments indicate that the primary mechanism of cartilage remodeling during fracture healing is through the action of MMPs, which can be expressed by a range of cell types and not solely osteoclasts. Critically, our in vitro evidence from articular sheep chondrocytes treated with either PD0325901 or AZD6244 showed an inhibition of Mmp13 expression.
The Effects of MEK Inhibition on Chondrocytes and Cartilage This study is the first to document the effects of either PD0325901 or AZD6244 on the growth plate and during fracture healing. Previous studies have successfully used PD0325901 at 20 to 50 mg/kg in mouse tumor xenograft models. We dosed mice with 10 to 25 mg/kg using a published oral delivery method that reduces gavage-induced stress. In a dose finding study, we had a 15 percent death rate after eight day treatment for mice that had undergone a fracture and treated with PD0325901 at 25 mg/kg/day data not shown. Therefore, we reduced the dose to 10 mg/kg, which has been suggested to be the clinically relevant equivalent in mice for PD0325901, and did not observe any toxicity. We observed an expansion of the hypertrophic zone in the MEKi treated mice, but the overall growth plate length was not changed. Therefore, the hypertrophic zone was expanded at the expense of the proliferative zone, suggesting that proliferative chondrocytes may be prematurely differentiating into hypertrophic chondrocytes. MEK inhibition using U0126 was shown to have direct impacts on hypertrophic chondrocytes. Inhibition of pERK using U0126 was associated with reduced hypertrophic chondrocyte apoptosis in culture, and a concomitant increase in the hypertrophic zone of the growth plate in mice. Notably however, PD0325901 has also been shown to increase serum phosphate levels, and this can be associated with increases in hypertrophic chondrocyte apoptosis.
Chondrocyte cultures revealed that MEKi could decrease Mmp13 expression, and this could underlie expansion of the hypertrophic zone. Mmp9 and Mmp13 knockout mice are reported to display an expansion of the hypertrophic zone of the growth plate, but this is also accompanied by a longer growth plate. Mechanistic studies have demonstrated that the effects of Mmp13 at the growth plate are predominantly due to a reduction in matrix turnover, rather than a change in hypertrophic chondrocyte differentiation or apoptosis. Some reductions in osteoclast parameters were noted at the level of the growth plate, but this is unlikely to perturb proliferative or hypertrophic chondrocyte differentiation. In Rankl minus minus mice that lack osteoclasts, the expansion of the growth plate and hypertrophic zone was associated with reduced matrix turnover, but chondrocyte differentiation was not impacted. As the MEKi treated mice do not show a change in growth plate length, we posit the hypertrophic zone expansion is not primarily caused by direct effects on MMP expression or osteoclasts. Most likely, these effects are secondary to direct effects of MEKi on chondrocyte differentiation.
In summary, we speculate that the impact of PD0325901 on the growth plate may be mediated through 1 premature differentiation of proliferating chondrocytes, 2 direct anti-apoptotic effects on hypertrophic chondrocytes, and 3 a protective effect from serum phosphate levels, which has been reported with PD0325901. These effects of MEKi treatment on the growth plate and on hypertrophic chondrocytes may warrant further examination in pediatric patients that are being administered these drugs in clinical trials.
Study Limitations Both of these MEKi compounds are currently in clinical development to treat cancer. The data presented from this study provide evidence that support an important role for the Ras–MAPK pathway in fracture healing and skeletal remodeling. We showed that PD0325901 and AZD6244 were capable of promoting cartilage formation during fracture healing, and delaying cartilage matrix resorption, resulting in altered endochondral ossification at the growth plate and fracture callus. The in vitro data suggests that MMP-13 inhibition may be the mechanism for delayed cartilage remodeling. The use of two MEK inhibitors at the same dose confirms the specificity of the effects on the MEK pathway, and allows a dosage comparison that is relevant for future studies.
We did not specifically assess angiogenesis, which is also an important process for endochondral ossification that may be affected by MEK inhibitors. The impact of these compounds on articular cartilage was not assessed either, though the in vitro data was highly informative. Finally, the long-term impact of MEKi compounds on the growth plate remains to be evaluated, since our results only reflect three weeks of treatment. The reversibility of the effects is promising, however.
Conclusions
These data demonstrate an important role for MEK signaling in the maintenance of cartilage and in endochondral bone repair. As MEKi compounds progress through clinical trials, evidence about how they impact the skeleton is essential, particularly in growing children or patients who sustain a fracture. Future studies may identify new ways to utilize this new class of drugs to modulate cartilage turnover and differentiation to improve bone and joint health.